
Download the USGS AI pack which contains patterns for geological mapping for Adobe illustrator. Instructions for using the pack are written below.
Download USGS_AI_Pack.zip
Instructions for using USGS AI Pack
This pack has been derived from:
There are a couple of different ways to use this pack:
Approach 1: Installing the swatches and using the reference.
- Open a new document in illustrator and bring up the swatches panel (Window>Swatches).
- Click on the small icon in the bottom left to choose swatches and go to Other Library.

This should put you in the folder that contains a folder of swatches (or the swatches folder itself) - Copy and Paste the “USGS_Swatches” folder you downloaded to the Swatches folder.
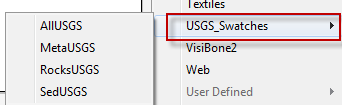
- Now when you click on the small icon in the bottom left of the Swatches Panel you will see USGS_Swatches in the list.
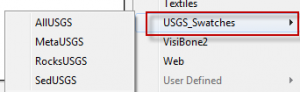
- Open up one of the Swatch sets. If you hover over the individual Swatches you will notice a number is displayed.
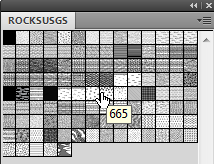
- Refer to the USGS_Swatches_reference.pdf to see which unit this refers to, or simply fill an object on the screen to view it. If you are using AllUSGS swatch refer to FGDCgeostdTM11A2_PattCh_poster.pdf
Approach 2: Opening the USGS pattern pages directly in illustrator
- In the folder called “USGS_pattern_pages” are four ai files. Open any one of these in illustrator.
- Select the pattern you want to use. Copy and paste this into your document (put it on the side of the page.)
- Use the eyedropper tool to apply this pattern to a shape. I.e select the shape, select the eyedropper then select the pattern.

54 comments on “USGS AI pack instructions and download link”
Thanks for this post – not sure how old the post is, but the download link is broken.
Just fixed the link. Should work now.
HI, does the link still function? I tried to download but I couldn’t
Thanks for your comment.
The download link is working for me.
We have found that sometimes the download does not work using a particular web browser. Have you tried with a different browser? eg Firefox or Chrome or Edge or …
I’m using it for my thesis.
I switch from Corel into AI because of the swatch.
Thank you so much for the AI swatch update!
No problem.
This is amazing. Very helpful.
It is missing dunham classifications for people who are doing logs of solely limestone and need to clarify between wackestone/packstone/grainstone/boundstone etc, but overall, this is unbelievably helpful. Thanks a bunch.
Luke, you’re a gem for posting these online and keeping the link active. They used to be really hard to find.
No problem.
Thanks for your sharing. One question can you tell me how to use illustrator to create a brush to use in a subduction zone?
thanks for your time.
I’m not really sure what you are asking. Brushes for Geological line symbols in illustrator are generally problematic.
I’ve lost count of how many time I have searched for the USGS swatch files via USGS and in the end I always find my way back here. Perfect. Thanks for keeping this updated!
No worries.
thank you thank you!!!!!!!!1
Very cool! Except I’m having an issue where even though I click the “fill” swatch, the “stroke” changes. Maybe this is an adobe issue, not an issue with the package?
Thought?
Thanks for posting this!
Hi, not sure what’s going wrong with that. Usually if you have the fill in the foreground then click on a pattern it will set the pattern as the fill. There’s a little icon to switch the stroke and fill. Does that fix it?
I’m having an issue about this pack.
How using this pallets in coreldraw platform?
Thanks
This is a great link indeed! I will use it for my dissertation. Though I have seen someone asking the question before; is it possible to get a swatch (or a brush) for structural features like thrust faults or subduction zones?
I did look into this. It’s a bit difficult in Illustrator as far as I can tell. You can use pattern brushes. But getting the regular placement of symbols doesn’t work so well.
Thank you! This is just what I was looking for, worked a treat!
Im running an older version of illustrator, CS4, and the program will import none of the swatches. Is there an older version of this I can access?
Hi, you should be able to open up the PDFs and use approach 2 above.
Very useful, thanks. The only thing I’d suggest is also supplying the swatches as SVG files for use as area fills in, say, QGIS.
Hi Rob, you can find them in inkscape svg format here:
https://blogs.otago.ac.nz/si-geology/resources/illustrationgraphics-resources/usgs-inkscape-pack-instructions-and-download-link/
I’d be interested to hear how you get on putting them in QGIS.
Note: I have to update all my inkscape templates due to a resolution change in inkscape.
Thank you for the upload! Is there anyway on AI to fill the pattern in with a color, or make the brushstroke larger on the patterns? It hard to see
Hi, yes there is. To get a colour in the background you can add an extra fill in the appearance dialog box behind the pattern. To make the pattern larger go to object transform and deselect all the options apart from pattern. There are also ways to completely modify the pattern such as pattern editor in adobe CS6 and later.
Gracias
I’m unable to use these swatches in AI CS4. I’ve tried a number of forum solutions but no luck. Do you have files specifically for AI CS4?
Never mind. I’m working it out.
Thanks a lot!
Impressive. Thank you so much
Impressive. Thanks a LOT!
many thanks
thank you very much admin for your important news to share with us. And more thanks for i get best idea read this article about this topics. many more days i was searched this topics, finally i got it.
Hi,
the link doesn’t work. Is there any way that you can get if fixed to work on Adobe Illustrator 2020?
Thank you
Hello, the download link works for me here but since you it doesn’t work for you perhaps this would be the place to try https://pubs.usgs.gov/tm/2006/11A02/
Good luck.
Gday, Fantastic work thank you very much.
question
I am doing a cross section and would like to align the symbology to the dip ie have the limestone texture on the dip angle. is there a way of doing this?
cheers
Hi, yes you can rotate the fill patterns from the USGS template (or any other fill).
Here’s how:
– choose the Edit paths nodes tool (N)
– click on the filled object that you want to modify
– look around the screen for the little circle that has just appeared (probably up and to the right of the object)
– move your cursor to that circle and drag the circle to interactively rotate the pattern
I hope that this works for you.
Stephen
Hi, I think the download link is broken. Thanks for the resource!
Thanks for your comment.
We have found that sometimes the download does not work using a particular web browser. Have you tried with a different browser? eg Firefox or Chrome or Edge or …
Unfortunately the link is broken again 🙁
Thanks for your comment.
We have found that sometimes the download does not work using a particular web browser. Have you tried with a different browser? eg Firefox or Chrome or Edge or …
I was able to download from the other page but when I download the .AI files they come as .ESP files. How do I get .AI file to be able to add to my library for use?
When you write ‘other page’ what page are you referring to?
Do you mean .eps ?
Adobe Illustrator should be able to open eps files.
Hi there,
Im using chrome and its not working? Should I use firefox or something else?
Thanks for your comment.
We have found that sometimes the download does not work using a particular web browser. Have you tried with a different browser? eg Firefox or Chrome or Edge or …
The download link doesn’t appear to be working, could you please try to upload a new link. Thanks!
Thanks for your comment.
The download link is working for me.
We have found that sometimes the download does not work using a particular web browser. Have you tried with a different browser? eg Firefox or Chrome or Edge or …
great tooll. a lot of thanks
Thank you so much for posting these!
Hello,
Thank you. Unfortunetly, the link is not working for me, doesn’t matter which web browser. Can you change the link ?
Updated http to https, hopefully that fixes it.
Great, succinct resource! The download link worked by copying and pasting into Chrome. Thanks!
I found what I was looking for! Thank you so much for providing these swatches/hatches for free!