
Research
Research
Our research is purely computational. We are primarily focussed on modelling catalysis of small molecule reactions on metallic catalysts. These are either extended metal surfaces or nanoparticles. We are also interested in structural determination of nanoparticles between ~100-5000 atoms in size. Please see below for more detail on some of our current and recent research projects.
Tools
We use a variety of software packages in our research projects. Click on the logos below to explore some of the tools that we use.




Research Topics
The Structure of Molybdenum Disulfide and Its Use as a Catalyst for the Hydrogen Evolution Reaction
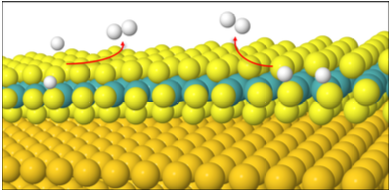
The hydrogen evolution reaction (HER) over a cheap catalyst such as molybdenum disulfide (MoS2) is part of a critical line of research dedicated to finding cheap, clean and sustainable fuel sources. It is important to model and investigate a range of conditions applied to these catalysts such that the most favourable conditions can be found. This involves developing an understanding of the mechanisms involved such that said conditions can be optimised efficiently to produce a more effective output of hydrogen, thus forming the basis of this research.
The main point of difference when investigating different conditions for MoS2 HER, is the interchanging of supporting catalyst (support) such as gold and graphene that rest under MoS2 changing its properties without being involved in the direct HER mechanisms. Another important variable condition is the coverage of hydrogen adsorbed to the MoS2 surface and how this may alter the favourability of proposed mechanisms.
It is accepted that the reactive sites of MoS2 occurs at the edges. Our research has involved finding the optimum sites on the edges for HER using mentioned supports as well as no support. Using these optimum sites, the mechanisms in which HER occurs are also a critical part of our current research.
It has been established that the first part of HER occurs by the Volmer reaction, where a hydrogen atom adsorbs onto the surface of MoS2. The second step of HER occurs by one or both of the following mechanisms (see above graphical representation of mechanisms):
Tafel, where two adsorbed hydrogens de-adsorb from the MoS2 surface to form dihydrogen.
Heyrovsky, where an adsorbed hydrogen de-adsorbs to react with a proton in solution to form dihydrogen.
Establishing which mechanism is favoured under different conditions is an area of research currently being investigated.
A secondary study involves the simulation of Raman spectra of several altered MoS2 structures in order to aid the assignment of experimentally observed signals.
Mechanism of NH3 synthesis on Ru nanoparticles
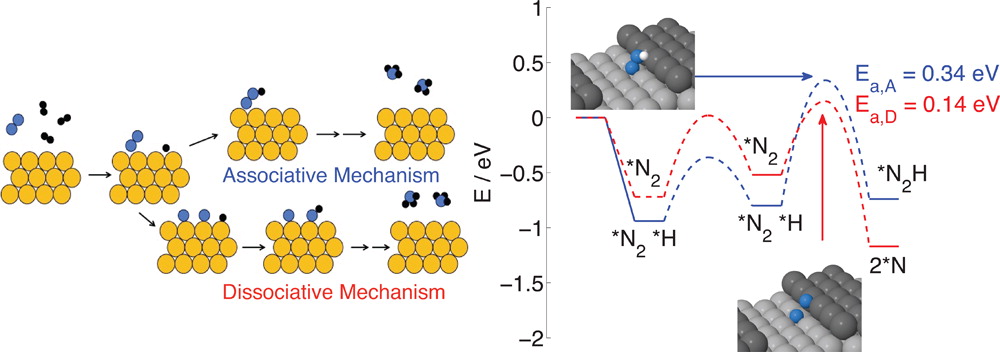
In collaboration with Assoc. Prof. Egill Skúlason at the University of Iceland. See Publication 13.
The Haber-Bosch process produces NH3 by passing gaseous H2 and atmospheric N2 over a metal catalyst at very high temperatures and pressures. Due to these harsh conditions, studying the mechanism by which ammonia forms has been a challenge. However, the mechanism needs to be understood for further optimisation of the catalyst, to improve efficiency and output.
There are two possible mechanisms; termed associative and dissociative (see figure above for a graphical representation of these). Previous research has indicated the major contribution to the rate is from the dissociative mechanism.
Recent advances in the field have renewed interest in studying both mechanisms, most importantly, the increased interest in nanoparticle catalysts, and the ability to use computational techniques to model the reaction. Nanoparticle catalysts offer dual benefits over extended metal surfaces: i) they have a high surface area to bulk ratio, and ii) they offer a high density of undercoordinated ‘active’ sites, such as corners, steps, and edges, which can contribute differently to the rate than planar sites (which dominate extended surfaces). In this research effort, we have been investigating both the associative and dissociative mechanisms on several different active sites which are potentially present on a Ru nanoparticle.
Structural determination of nanoparticles of catalytic interest
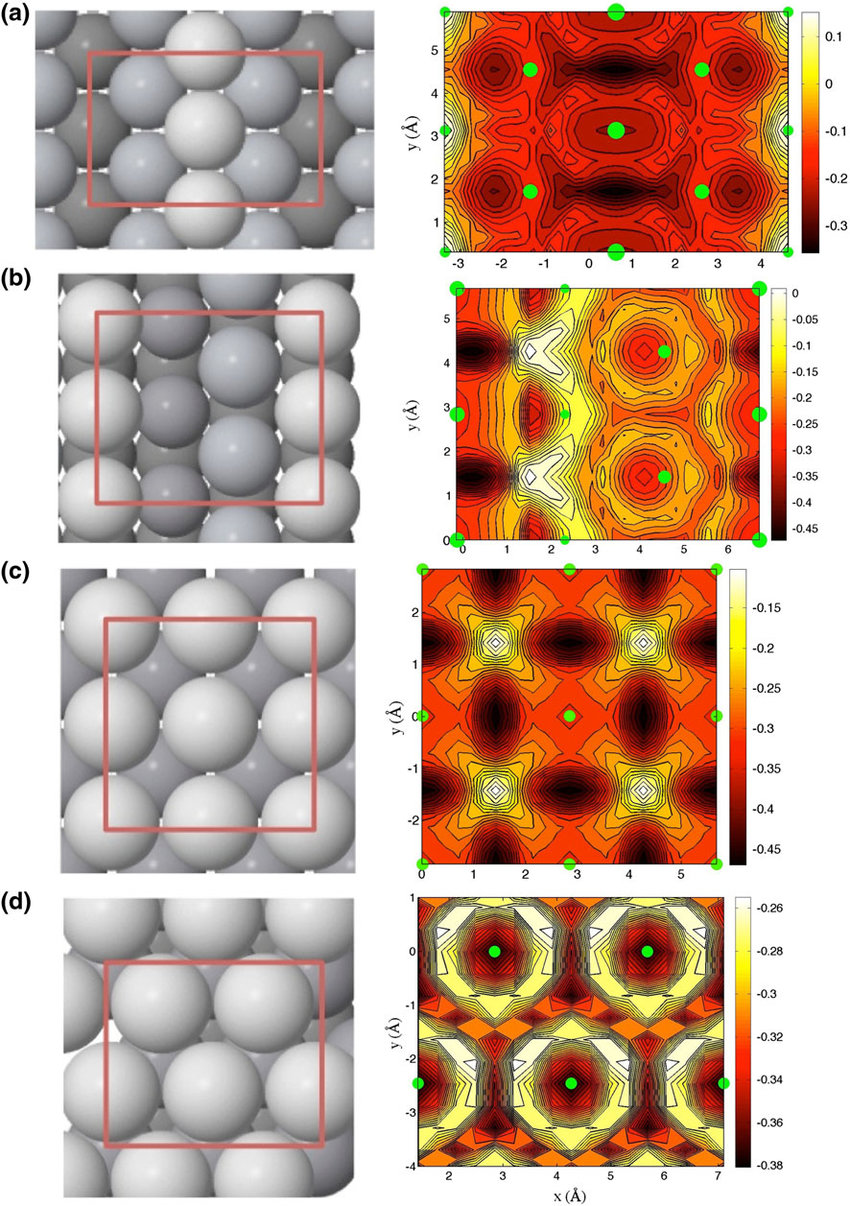
See Publication 10.
Nanoparticles make ideal catalysts as they can show drastically different properties to the bulk, due to the high surface area available for reaction and the high density of undercoordinated sites, such as steps, edges and corner sites. These undercoordinated sites are usually significantly more active than flat active sites and often dominate the catalytic activity of the entire nanoparticle.
We have recently investigated the catalytic activity towards H2 formation of two different types of edge sites that may be present in an FCC Pt nanoparticle. We found striking results: these two types of edge sites differ significantly in reactivity, by around 5 orders of magnitude at room temperature! This indicates that to accurately model the catalytic activity of nanoparticles, we must understand the likely shape of the particles and the types of undercoordinated sites present.
Predicting nanoparticle shape corresponds to a global optimisation problem. For most nanoparticles of interest (>100 atoms) this is a prohibitively difficult task. Thus, the focus in our group is to conduct broad screening of nanoparticle shapes to attempt to (i) elucidate trends in shape dominance with size and (ii) hone in on size regions that are likely to yield nanoparticles that exhibit high catalytic activity. We are currently studying Au and Pt nanoparticles in collaboration with Prof. Richard Palmer of U. Birmingham as well as Cu and Pd nanoparticles with Dr. Andreas Pedersen of ETH Zurich.
Currently, a genetic algorithm developed by Geoffrey Weal is being used for global optimisation of nanoparticles. This algorithm incorporates ideas of Darwinian evolution (e.g. survival of the fittest) to locate the lowest energy structure. Nanoparticles can be ‘mated’ together to generate new structures, or can undergo mutations. Currently we are seeking to improve the algorithm’s efficiency by tweaking the fitness operator, which determines which nanoparticles are culled from the population. You can find the Garden Genetic Algorithm (GGA) currently being developed on our Github page (https://github.com/GardenGroupUO). Instructions to using the GGA can be found at (https://gga.readthedocs.io/).
Electrochemical NH3 synthesis
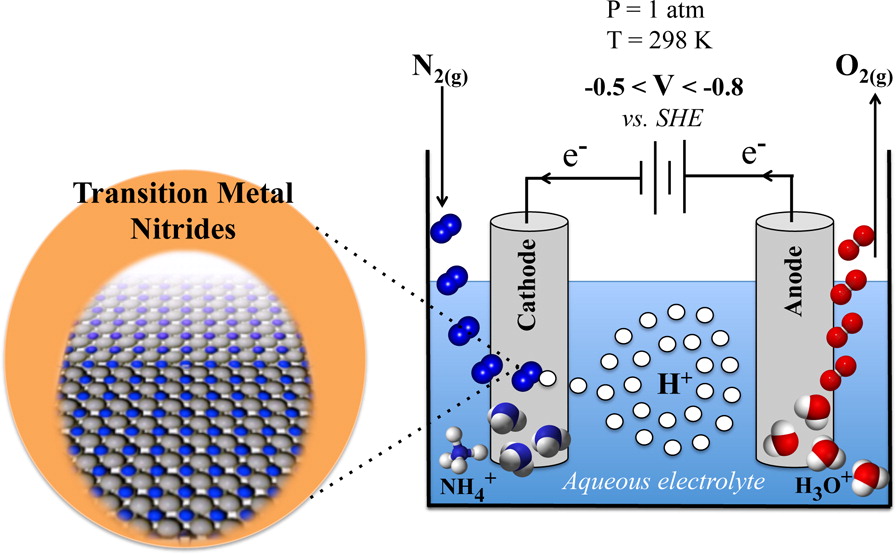
In collaboration with Assoc. Prof. Egill Skúlason at the University of Iceland. See Publications 12 and 14.
The production of NH3 by the Haber-Bosch process operates at high temperatures and pressures and requires gaseous H2 as a reactant. In contrast, the enzyme nitrogenase produces NH3 at ambient conditions, using solvated protons and electrons, rather than H2(g). Given the mild operating conditions and possibility of using renewable electricity to drive the reaction, it is an alluring prospect to develop an analogous man-made electrochemical method of synthesising NH3.
We have recently been investigating transition metal nitrides as possible electrode materials for electrochemical NH3 formation. We have identified four potential nitride candidates that satisfy the following necessary conditions: (i) They form NH3 with a low applied bias and at a reasonable rate; (ii) They are stable with respect to nitrogen leaking from the material; (iii) They are stable with respect to poisoning in an electrochemical environment; (iv) They are stable with respect to decomposition with an applied bias; (v) They are unlikley to form oxides. Our promising nitrides are currently being grown and tested for electrochemical activity.