
Professor Tony Blakely, Associate Professor Nick Wilson, Professor John Attia (University of Newcastle and Hunter Medical Research Institute, Australia)
 Many observational studies suggest that moderate alcohol consumption reduces cardiovascular disease (CVD) risk. The so-called J-shaped curve. We summarised the ‘state of play’ on this topic at PHE Blog in early 2014. In the last few weeks a large ‘Mendelian randomisation’ study (like a genetically determined RCT of alcohol consumption – a study design with much potential for identifying causal (as opposed to confounded) associations) has been published suggesting that any alcohol consumption increases CVD. If true, this means the overall health harm from alcohol consumption in our society is even higher than currently thought. If so it would make cost-effective interventions such as alcohol taxation even more justified.
Many observational studies suggest that moderate alcohol consumption reduces cardiovascular disease (CVD) risk. The so-called J-shaped curve. We summarised the ‘state of play’ on this topic at PHE Blog in early 2014. In the last few weeks a large ‘Mendelian randomisation’ study (like a genetically determined RCT of alcohol consumption – a study design with much potential for identifying causal (as opposed to confounded) associations) has been published suggesting that any alcohol consumption increases CVD. If true, this means the overall health harm from alcohol consumption in our society is even higher than currently thought. If so it would make cost-effective interventions such as alcohol taxation even more justified.
In this blog, we explain and critique this latest study in the British Medical Journal. Early in 2014 we concluded that:
“There may be a benefit for cardiovascular disease prevention from moderate alcohol use – in fact we wager there probably is. But its true benefit may be overstated by our current ‘best evidence’ due to the unresolvable biases.”
Based on this latest study – which is useful but far from definitive – we shift slightly our expert opinion to “… we wager there possibly is, …”. That is, the evidence base for cardio-protective effects of moderate alcohol consumption is weakening – but has not vaporised.
So what is this Mendelian randomisation study design? In a nutshell, very clever and with the (possibility) of being much better at identifying causal associations than ‘traditional’ observational studies. The trick is to use your random assignment of genes at birth as a type of RCT – there is a random chance you will inherit the variant in the alcohol dehydrogenase gene that makes you less likely to drink alcohol. This is because the variant enzyme metabolises alcohol to acetaldehyde 40 times faster than the usual enzyme; acetaldehyde is a toxic metabolite that causes flushing, sweating, an accelerated heart rate and nausea and vomiting – which leads to drinking less alcohol. That is, by identifying those people with and without this genetic variant, there is a random (and therefore unconfounded) variation of alcohol consumption. Because of attenuation of effects, these studies need to be massive. This current study was a meta-analysis (pooling multiple individual studies) of 262,000 people.
What did the study find? For people with this genetic difference in alcohol metabolism, alcohol consumption was less (as theoretically expected) and the rate of CVD was less – at all levels of alcohol consumption. That is, this study did not find evidence of a J-shaped association of alcohol with CVD – rather, each additional unit of alcohol appeared to increase the risk of CVD.

A diagram from the BMJ showing how Mendelian randomisation works like an RCT (source: Burgess et al, 2012, doi: http://dx.doi.org/10.1136/bmj.e7325)
Do we fully trust this study’s conclusions? No – it is just one (albeit large and ‘good’) study. Consistent with a cautious editorial by Glymour (one of the new cohort of extremely good epidemiologists in matters of causal inference), we find a couple of threats to the assumptions of a good Mendelian randomisation study. First, the genetic variant not only affects alcohol consumption, but also the metabolites of alcohol – so maybe it is the metabolites of alcohol that are having the effect, not alcohol per se? Second, there should be no association of the genetic variant with CVD among the non-alcohol drinkers – otherwise one suspects that the genetic variant is also effecting CVD through pathways other than just alcohol, and this would tend to overestimate the effect of alcohol. This ‘non-association’ did not hold; in non-drinkers, the genetic variant was significantly associated with a number of intermediates that affect CVD, including systolic blood pressure, interleukin-6, BMI, and non-HDL cholesterol. Thirdly, it is customary to “close the triangle”, i.e. having estimated the effect of the gene on the exposure (alcohol), and the gene on the outcome (CVD), one estimates the “expected” effect of exposure on outcome and compares this with that actually observed. This last step is not provided in the paper; there is no calculation of the expected effect of reduced alcohol on CVD and no comparison to that actually measured across all the studies.
Nevertheless, the overall pattern of findings was consistent with a deleterious effect of alcohol. As expected, the genetic variant had no association with coronary heart disease among non-drinkers (a relative risk of 0.98, 95% confidence interval 0.90 to 1.07), but did show an association among drinkers (0.86, 0.78 to 0.94).
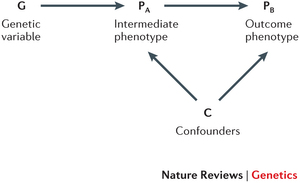
A causal diagram of Mendelian randomisation. The idea is that the genetic variable influences a risk factor (e.g. alcohol consumption) and nothing else. Thus, the genetic variable has no influence or association with ‘outcome phenotype’ (e.g. coronary heart disease) other than through the ‘intermediate phenotype’ of interest. But the association of the intermediate phenotype with outcome phenotype can still be confounded (as shown).
So what? This study further increases the evidence against a meaningful J-shape association of alcohol with CVD, but it does not entirely rule it out – this study is a not a ‘top drawer’ Mendelian randomisation study. Yet given this and other evidence (as we reviewed before) it is probably not scientifically appropriate for public health practitioners, policy makers, and the media to continue to state that moderate alcohol consumption is likely to be good for heart health. Rather, such a contention is quite speculative and the balance of evidence for alcohol and health is very much negative overall (given the proven risks of: injury, addiction, liver disease, and some cancers – including breast cancer).
Smart policy makers have a tricky job balancing the benefits society gets from alcohol (pleasure, social lubrication and use in celebrations) with the health harms and societal harm (crime and violence etc). Nevertheless, the current evidence adds support for society continuing to use such cost-effective instruments as alcohol taxes and regulations around marketing and access to alcohol.
I think all of us who drink at least a little alcohol have a personal interest in this which might induce some readers’ bias into acceptance of results. There are indeed some tenuous links in this study but it does add to the confounding argument. Another recent study shows that even low consumption of alcohol increases the risk of atrial fibrillation (http://content.onlinejacc.org/article.aspx?articleID=1889060) as with hypertension although, again, a genetic link between alcohol appetite/tolerance and risk factors or disease is an intriguing thought.
There is much potential in using Mendelian-type randomization techniques, I am thinking particularly of nutrition and the doomed nutrient in-mortality out current models. There is potential to construct complex webs of causation utilizing post-translational known modifications in a given micronutrient pathway, blood parameters of associated metabolites that reflect organ compensations that may in themselves not be significant, which would give much more power to any association that tries to lead from genetic isoforms to disease outcome. The other consideration is the differential weighting of each variable. It is not good enough to minimise unknowns to tidy up the statistics.
‘the variant enzyme metabolises alcohol to acetaldehyde 40 times faster than the usual enzyme’
This is a misleading partially-true statement. It leads to the notion that the foetus of a pregnant woman in the first trimester may be 40 times more likely to have alcohol syndrome than ‘normal’. It quotes extremes, an either/or scenario that does not exist, though is easily fit into statistical models. The optimum functioning of dehydrogenase enzymes also depends on nutrient co-factor supply, whole body status, and many epigenetic cues to gene translation and expression. People have varying capacities and inborn programming for receptor numbers and functioning. These can be grouped into slow/fast metabolisers for whatever reason, with genetic isoforms playing their part.